NALgen Analysis Pt.0
Non-negative matrix factorization, as implemented in the LEA package, is a case of unsupervised machine learning; LEA uses least-squares estimates of ancestry coefficients.
Presented below are the reference results that the 20-population-subset results will be compared against. The original landscapes used to simulate the genetic data, as well as population structure results obtained from these complete genetic datasets, will be used in evaluating the NALgen method.
Multivariate analyses: Using machine learning algorithms to infer population structure
Loading the environmental data
library(raster)
library(virtualspecies)
setwd("C:/Users/chazh/Documents/Research Projects/Reticulitermes/Phylogeography/Geo_Analysis/Data/EnvData/bioclim_FA/east coast/Finalized/current")
fn <- list.files(pattern=".asc")
fa_stk <- stack()
for(i in 1:length(fn)) fa_stk=addLayer(fa_stk,raster(fn[i]))
#names for plot titles
fa_stk_names <- c("Annual Temperature Range", "Dry-Season Precipitation", "Summer Temperature", "Wet-Season Precipitation")
Loading the genetic data
library(adegenet)
setwd("C:/Users/chazh/Documents/Research Projects/Reticulitermes/NALgen_Analysis/Multivariate/")
#below I define the xyFromRC function: includes a scenario when genind and rastr don't have the same dimensions
#currently (for my purposes here it's fine) only spits out error when aspect ratio is not the same, but proceeds the same anyway
#need to fix to apply to other situations
xyFromRC <- function(genind, rastr){
coord <- genind$other
row.no <- coord[,1]+1
col.no <- coord[,2]+1
dim.genind.row <- 26 #dim of simulated landscape
dim.genind.col <- 34 #dim of simulated landscape
dim.rast.row <- dim(rastr)[1]
dim.rast.col <- dim(rastr)[2]
if(dim.rast.col/dim.genind.col == dim.rast.row/dim.genind.row){
print("raster and genind are the same aspect ratio. proceed.")
}
else{
print("error! raster and genind are NOT the same aspect ratio. proceed ANYWAY.")
}
rastr <- aggregate(rastr, dim.rast.col/dim.genind.col)
cell.no <- cellFromRowCol(rastr, row=row.no, col=col.no)
coord <- xyFromCell(rastr, cell.no)
as.data.frame(coord)
}
#use already aggregated fa_stk to make this go faster
fa_stk.agg <- aggregate(fa_stk, dim(fa_stk)[1]/26)
#folders with full simulated data (not 20-pop subsets)
full_m0.1_lclp <- "C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_1/Simulated_Landscapes/500neutral+50selectedSNPs/Low_Complexity_Landscapes/landscapeR/Low_Permeability"
full_m0.1_lchp <- "C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_1/Simulated_Landscapes/500neutral+50selectedSNPs/Low_Complexity_Landscapes/landscapeR/High_Permeability"
full_m0.1_hclp <- "C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_1/Simulated_Landscapes/500neutral+50selectedSNPs/High_Complexity_Landscapes/virtualspecies/Low_Permeability"
full_m0.1_hchp <- "C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_1/Simulated_Landscapes/500neutral+50selectedSNPs/High_Complexity_Landscapes/virtualspecies/High_Permeability"
full_m0.5_lclp <- "C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_5/Simulated_Landscapes/500neutral+50selectedSNPs/Low_Complexity_Landscapes/landscapeR/Low_Permeability"
full_m0.5_lchp <- "C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_5/Simulated_Landscapes/500neutral+50selectedSNPs/Low_Complexity_Landscapes/landscapeR/High_Permeability"
full_m0.5_hclp <- "C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_5/Simulated_Landscapes/500neutral+50selectedSNPs/High_Complexity_Landscapes/virtualspecies/Low_Permeability"
full_m0.5_hchp <- "C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_5/Simulated_Landscapes/500neutral+50selectedSNPs/High_Complexity_Landscapes/virtualspecies/High_Permeability"
setwd(full_m0.1_lclp)
full_m0.1_lclp_n.s <- read.genepop("lclp.GENEPOP.gen")
full_m0.1_lclp_n.s$other <- read.table("lclp.GENEPOP.PopCoor.txt", sep=" ",header=F)
colnames(full_m0.1_lclp_n.s$other)=c("x","y")
full_m0.1_lclp_n.s$other <- xyFromRC(full_m0.1_lclp_n.s, fa_stk.agg)
setwd(full_m0.1_lchp)
full_m0.1_lchp_n.s <- read.genepop("lchp.GENEPOP.gen")
full_m0.1_lchp_n.s$other <- read.table("lchp.GENEPOP.PopCoor.txt", sep=" ",header=F)
colnames(full_m0.1_lchp_n.s$other)=c("x","y")
full_m0.1_lchp_n.s$other <- xyFromRC(full_m0.1_lchp_n.s, fa_stk.agg)
setwd(full_m0.1_hclp)
full_m0.1_hclp_n.s <- read.genepop("hclp.GENEPOP.gen")
full_m0.1_hclp_n.s$other <- read.table("hclp.GENEPOP.PopCoor.txt", sep=" ",header=F)
colnames(full_m0.1_hclp_n.s$other)=c("x","y")
full_m0.1_hclp_n.s$other <- xyFromRC(full_m0.1_hclp_n.s, fa_stk.agg)
setwd(full_m0.1_hchp)
full_m0.1_hchp_n.s <- read.genepop("hchp.GENEPOP.gen")
full_m0.1_hchp_n.s$other <- read.table("hchp.GENEPOP.PopCoor.txt", sep=" ",header=F)
colnames(full_m0.1_hchp_n.s$other)=c("x","y")
full_m0.1_hchp_n.s$other <- xyFromRC(full_m0.1_hchp_n.s, fa_stk.agg)
setwd(full_m0.5_lclp)
full_m0.5_lclp_n.s <- read.genepop("lclp.GENEPOP.gen")
full_m0.5_lclp_n.s$other <- read.table("lclp.GENEPOP.PopCoor.txt", sep=" ",header=F)
colnames(full_m0.5_lclp_n.s$other)=c("x","y")
full_m0.5_lclp_n.s$other <- xyFromRC(full_m0.5_lclp_n.s, fa_stk.agg)
setwd(full_m0.5_lchp)
full_m0.5_lchp_n.s <- read.genepop("lchp.GENEPOP.gen")
full_m0.5_lchp_n.s$other <- read.table("lchp.GENEPOP.PopCoor.txt", sep=" ",header=F)
colnames(full_m0.5_lchp_n.s$other)=c("x","y")
full_m0.5_lchp_n.s$other <- xyFromRC(full_m0.5_lchp_n.s, fa_stk.agg)
setwd(full_m0.5_hclp)
full_m0.5_hclp_n.s <- read.genepop("hclp.GENEPOP.gen")
full_m0.5_hclp_n.s$other <- read.table("hclp.GENEPOP.PopCoor.txt", sep=" ",header=F)
colnames(full_m0.5_hclp_n.s$other)=c("x","y")
full_m0.5_hclp_n.s$other <- xyFromRC(full_m0.5_hclp_n.s, fa_stk.agg)
setwd(full_m0.5_hchp)
full_m0.5_hchp_n.s <- read.genepop("hchp.GENEPOP.gen")
full_m0.5_hchp_n.s$other <- read.table("hchp.GENEPOP.PopCoor.txt", sep=" ",header=F)
colnames(full_m0.5_hchp_n.s$other)=c("x","y")
full_m0.5_hchp_n.s$other <- xyFromRC(full_m0.5_hchp_n.s, fa_stk.agg)
Unsupervised classification: Inferring genetic clusters using non-negative matrix factorization (LEA package)
library(ggsci)
library(LEA)
library(mapplots)
library(maps)
futr <- pal_futurama()
schwifty <- pal_rickandmorty()
futr.cols <- colorRampPalette(futr(12)[c(12,11,3,7,8,6,1,2)])
schwifty.cols <- colorRampPalette(schwifty(12)[c(3,12,4,6,1,9,8,2)])
futrschwift.cols <- colorRampPalette(c("gray15", schwifty(12)[3], futr(12)[11], schwifty(12)[c(1,9)], futr(12)[c(8,6,2)]))
cols <- futrschwift.cols(12)
#plot parameters
par(mfrow=c(2,2), fg="gray50", pty='m', bty='o', mar=c(4,4,4,4), cex.main=1.15, cex.axis=1.05, cex.lab=1.1)
geninds <- list(full_m0.1_lclp_n.s,
full_m0.1_lchp_n.s,
full_m0.1_hclp_n.s,
full_m0.1_hchp_n.s,
full_m0.5_lclp_n.s,
full_m0.5_lchp_n.s,
full_m0.5_hclp_n.s,
full_m0.5_hchp_n.s)
genind_names <- c("full_m0.1_lclp_n.s",
"full_m0.1_lchp_n.s",
"full_m0.1_hclp_n.s",
"full_m0.1_hchp_n.s",
"full_m0.5_lclp_n.s",
"full_m0.5_lchp_n.s",
"full_m0.5_hclp_n.s",
"full_m0.5_hchp_n.s")
ls_names <- c("LCLP landscape (m = 0.1)",
"LCHP landscape (m = 0.1)",
"HCLP landscape (m = 0.1)",
"HCHP landscape (m = 0.1)",
"LCLP landscape (m = 0.5)",
"LCHP landscape (m = 0.5)",
"HCLP landscape (m = 0.5)",
"HCHP landscape (m = 0.5)")
lea.dir <- c(
"C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_1/Simulated_Landscapes/500neutral+50selectedSNPs/Low_Complexity_Landscapes/landscapeR/Low_Permeability/",
"C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_1/Simulated_Landscapes/500neutral+50selectedSNPs/Low_Complexity_Landscapes/landscapeR/High_Permeability/",
"C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_1/Simulated_Landscapes/500neutral+50selectedSNPs/High_Complexity_Landscapes/virtualspecies/Low_Permeability/",
"C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_1/Simulated_Landscapes/500neutral+50selectedSNPs/High_Complexity_Landscapes/virtualspecies/High_Permeability/",
"C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_5/Simulated_Landscapes/500neutral+50selectedSNPs/Low_Complexity_Landscapes/landscapeR/Low_Permeability/",
"C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_5/Simulated_Landscapes/500neutral+50selectedSNPs/Low_Complexity_Landscapes/landscapeR/High_Permeability/",
"C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_5/Simulated_Landscapes/500neutral+50selectedSNPs/High_Complexity_Landscapes/virtualspecies/Low_Permeability/",
"C:/Users/chazh/Documents/Research Projects/Reticulitermes/Simulations/popRange/m_0_5/Simulated_Landscapes/500neutral+50selectedSNPs/High_Complexity_Landscapes/virtualspecies/High_Permeability/"
)
bestK <- c()
for (i in 1:8){
genind.obj <- geninds[[i]]
genotypes <- genind.obj$tab
coords <- genind.obj$other
##needed when running pop structure inference
#setwd(lea.dir[i])
#write.table(t(as.matrix(genotypes)), sep="", row.names=F, col.names=F, paste0(genind_names[i], ".geno"))
#geno <- read.geno(paste0(genind_names[i],".geno"))
setwd(paste0(lea.dir[i],"LEA"))
source("POPSutilities.r")
setwd(lea.dir[i])
reps <- 5
maxK <- 15
##needed when running pop structure inference
#snmf.obj <- snmf(paste0(genind_names[i],".geno"), K=1:maxK, repetitions=reps, project="new",
# alpha=10, iterations=100000,
# entropy=TRUE, percentage=0.25)
#plot(snmf.obj, cex=1.2, col="lightblue", pch=19)
#loading saved files from previous LEA run
snmf.obj <- load.snmfProject(paste0(genind_names[i],".snmfProject"))
#determining best K and picking best replicate for best K
ce <- list()
for(k in 1:maxK) ce[[k]] <- cross.entropy(snmf.obj, K=k)
ce.K <- c()
for(k in 1:maxK) ce.K[k] <- min(ce[[k]])
diff <- ce.K[-1] - ce.K[-maxK]
slope <- exp(-diff) - 1
#K is selected based on the smallest slope value in the upper quartile
best.K <- min(which(slope <= quantile(slope)[4]))
best.run <- which.min(ce[[best.K]])
bestK[i] <- best.K
#plot(ce.K, pch=21, cex=1.1, bg="red3", ylab="Cross-entropy", xlab="K")
#plot(diff, pch=21, cex=1.1, bg="red3", ylab="Difference", xlab="K")
#plot(slope, pch=21, cex=1.1, bg="red3", ylab="Slope", xlab="K")
barchart(snmf.obj, K=best.K, run=best.run,
border=NA, space=0, col=futrschwift.cols(best.K),
xlab="Individuals", ylab="Ancestry proportions",
main=paste("Ancestry:", ls_names[i]))
qmatrix <- Q(snmf.obj, K=best.K, run=best.run)
grid <- createGrid(xmin(fa_stk), xmax(fa_stk),
ymin(fa_stk), ymax(fa_stk), 260, 340)
constraints <- NULL
shades <- 10
futrschwift.gradient <- colorRampPalette(futrschwift.cols(best.K))
grad.cols <- futrschwift.gradient(shades*best.K)
ColorGradients_bestK <- list()
for(j in 1:best.K){
k.start <- j*shades-shades+1
k.fin <- j*shades
ColorGradients_bestK[[j]] <- c("gray95", grad.cols[k.start:k.fin])
}
maps(qmatrix, coords, grid, constraints, method="max",
colorGradientsList=ColorGradients_bestK,
main=paste("Mapped ancestry:", ls_names[i]),
xlab="Longitude", ylab="Latitude", cex=.4)
map(add=T, interior=F)
}
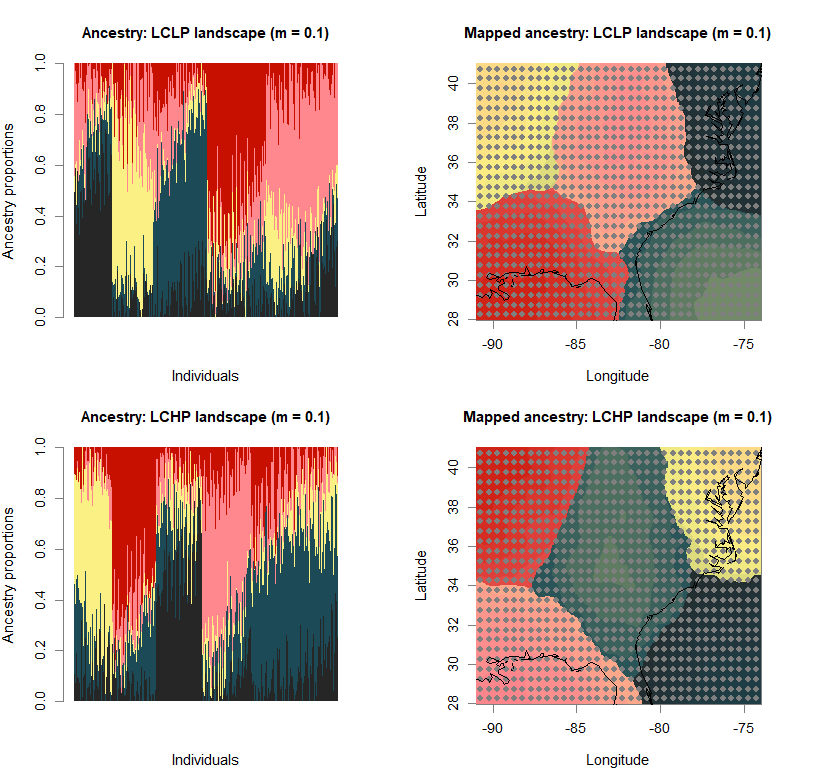
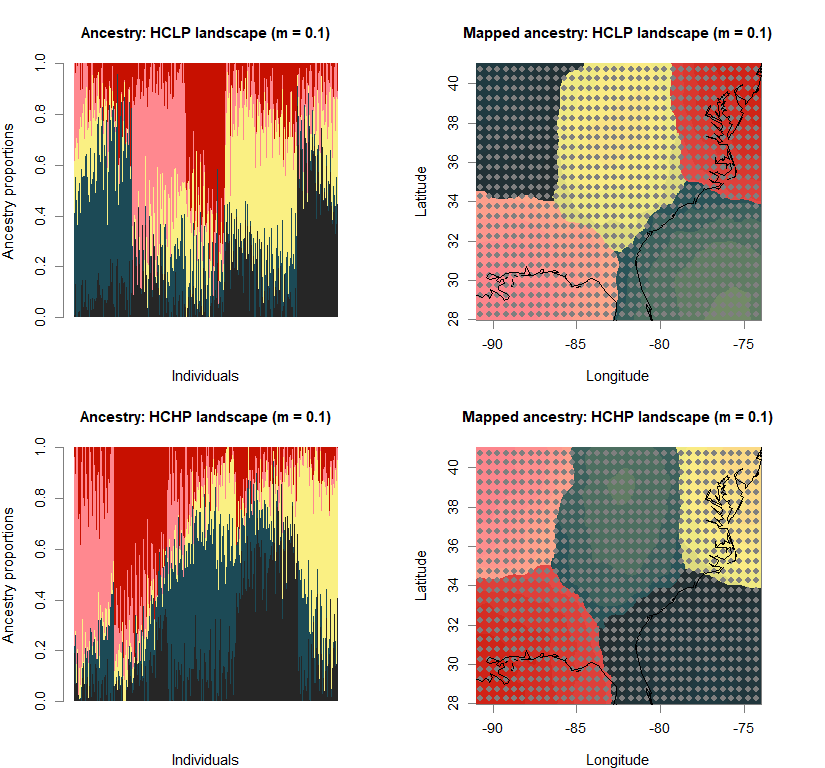
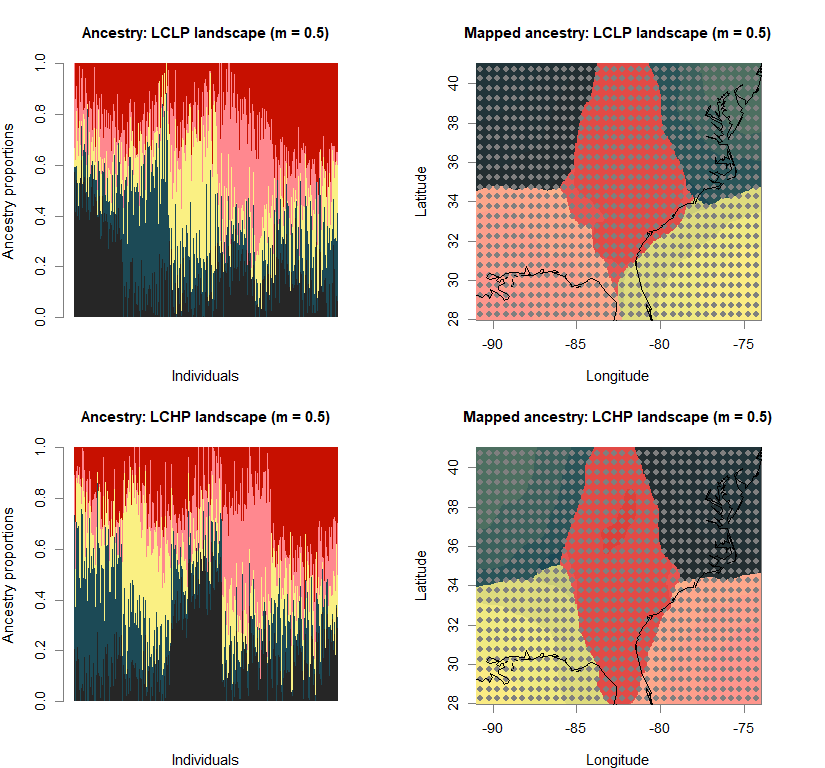
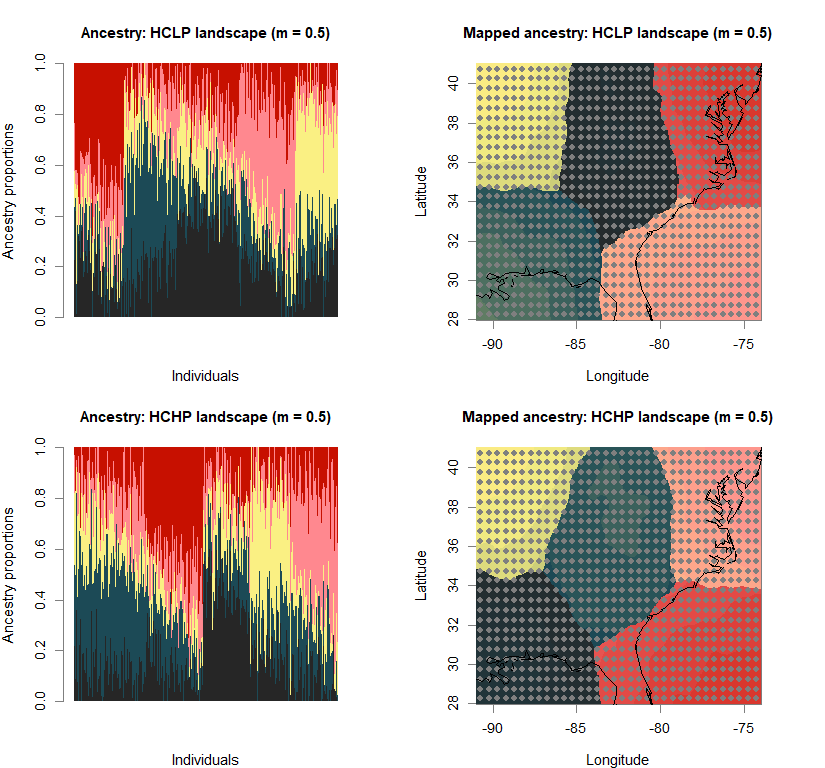
names(bestK) <- ls_names
print(bestK)
## LCLP landscape (m = 0.1) LCHP landscape (m = 0.1) HCLP landscape (m = 0.1)
## 5 5 5
## HCHP landscape (m = 0.1) LCLP landscape (m = 0.5) LCHP landscape (m = 0.5)
## 5 5 5
## HCLP landscape (m = 0.5) HCHP landscape (m = 0.5)
## 5 5
